

С 30 апреля по 4 мая проходила моя любимая ежегодная конференция PEGS Boston. На протяжении 14 лет в дизайне логотипа и всех сопутствующих материалов этой конференции используется изображение одного и того же белка, и вот уже 13 лет подряд организаторы конференции задают вопрос: что это за белок? В этом году команда ДВБ впервые ответила на этот вопрос. И теперь главный вопрос: как?
В первую очередь давайте обратим внимание на изображение. Это cartoon-представление белка, в котором альфа-спирали заменены на цилиндры. В PyMOL подобного поведения можно добиться следующими настройками:
show_as cartoon, all
set cartoon_fancy_helices, 0
set cartoon_cylindrical_helices, 1
Также белок покрашен в режиме спектра, распределенного по длине последовательности. Самая первая аминокислота покрашена в фиолетовый, а последняя в красный. Делается это еще одной простой командой:
spectrum count, rainbow
Теперь мы можем превратить стандартное полноатомное представление
в симпатичный вид а-ля PEGS
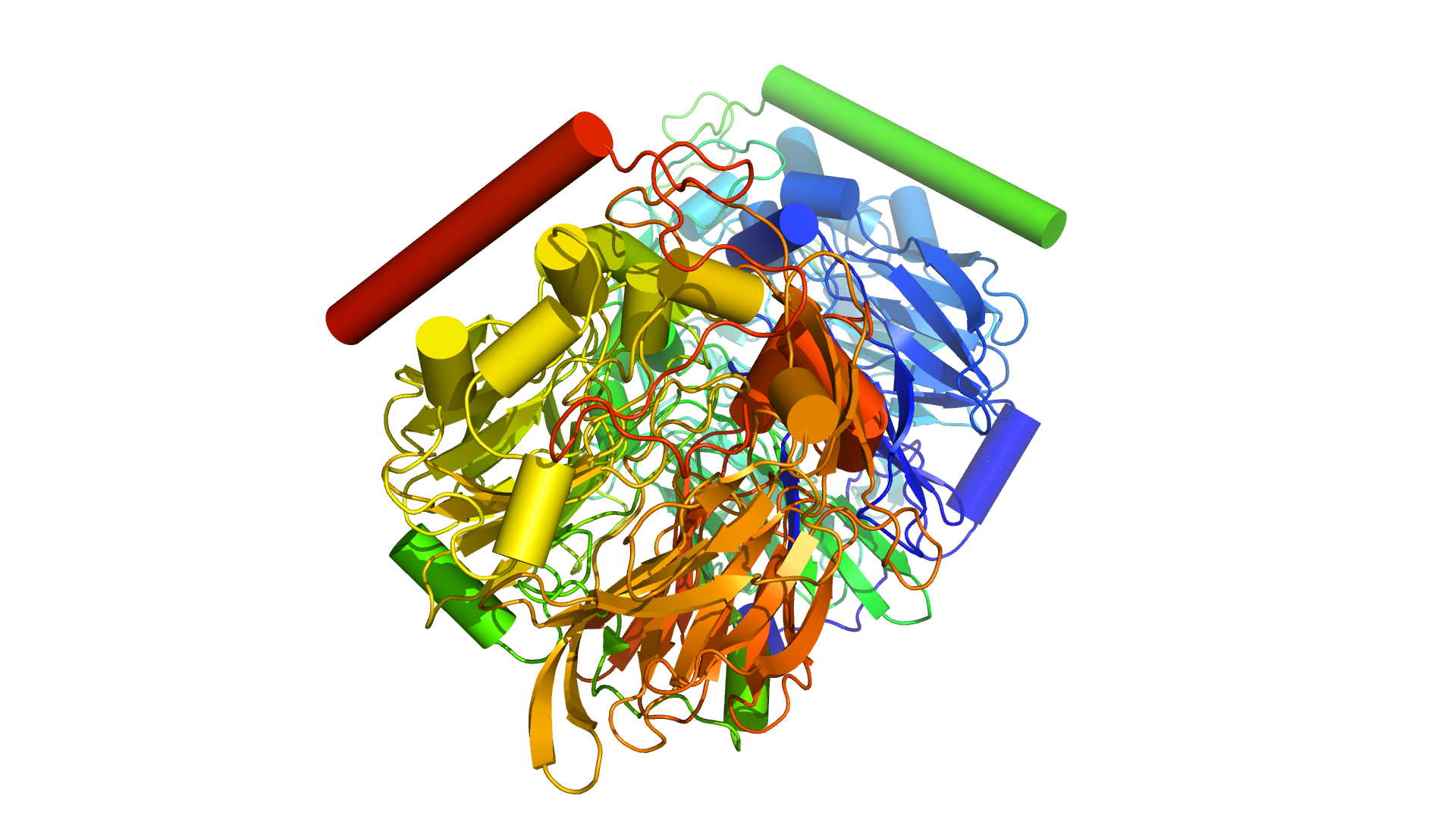
Наша первая идея состояла в том, что можно нагенерировать изображений из Protein DataBank и сравнить их с нашим изображением, например, 2D-Хэммингом. Впоследствии мы отказались от этой идеи (а зря!), но скрипт генерации у нас появился:
from pymol import cmd
import os
DIR = '/path/to/locate/pdbs'
IMG = '/path/to/store/images'
if not os.path.exists(IMG):
os.mkdir(IMG)
for ent in os.listdir(DIR):
filename = os.path.join(IMG, "%s.png" % ent)
if os.path.exists(filename):
continue
cmd.load(os.path.join(DIR, ent))
cmd.show_as('cartoon', 'all')
cmd.set('cartoon_fancy_helices', 0)
cmd.set('cartoon_cylindrical_helices', 1)
cmd.spectrum('count', 'rainbow')
cmd.orient()
cmd.save(filename)
cmd.reinitialize()
Запуск через консольный режим PyMOL делается тоже просто:
pymol -qc make_images.py
Отказались мы от этой идеи по причине того, что актуальный PDB имеет уж слишком много записей. На момент написания этого поста статистика следующая:
| Experimental Method | Proteins | Nucleic Acids | Protein/NA Complex | Other | Total |
|---|---|---|---|---|---|
| X-Ray | 117481 | 1919 | 6011 | 10 | 125421 |
| NMR | 10708 | 1243 | 249 | 8 | 12208 |
| Electron Microscopy | 1546 | 31 | 542 | 0 | 2119 |
| Other | 215 | 4 | 6 | 13 | 238 |
| Multi Method | 116 | 4 | 2 | 1 | 123 |
| Total | 130066 | 3201 | 6810 | 32 | 140109 |
В этом нам очень помогло, что Саша Надолинский узнал автора изображения и нашел на его сайте картинку высокого разрешения. Из нее мы пришли к выводу, что искать требуется белок с одной цепью, в состав которого входит не менее 30 альфа-спиралей, не менее 17 из которых имею длину 5 и менее аминокислот. Также мы обратили внимание, что в начале цепи идет альфа-спираль, так что мы решили добавить это знание в наши фильтры. Единственное, мы не были уверены, что раскраска цепи каноническая от синего к красному, поскольку помимо классического режима rainbow существует также rainbow_rev, в котором все ровно наоборот. С другой стороны белка также присутствовала спираль, но не с самого начала. В связи с этим мы решили перестраховаться и сказать, что 3 позиция должна покрываться спиралью.
Разумеется, главным критерием отсева была дата: первый PEGS проводился в 2005 году, а значит белок должен был быть кристаллизован ранее. Если посмотреть на график роста размера Protein DataBank, видно, что это действительно мощнейший фильтр из всех.
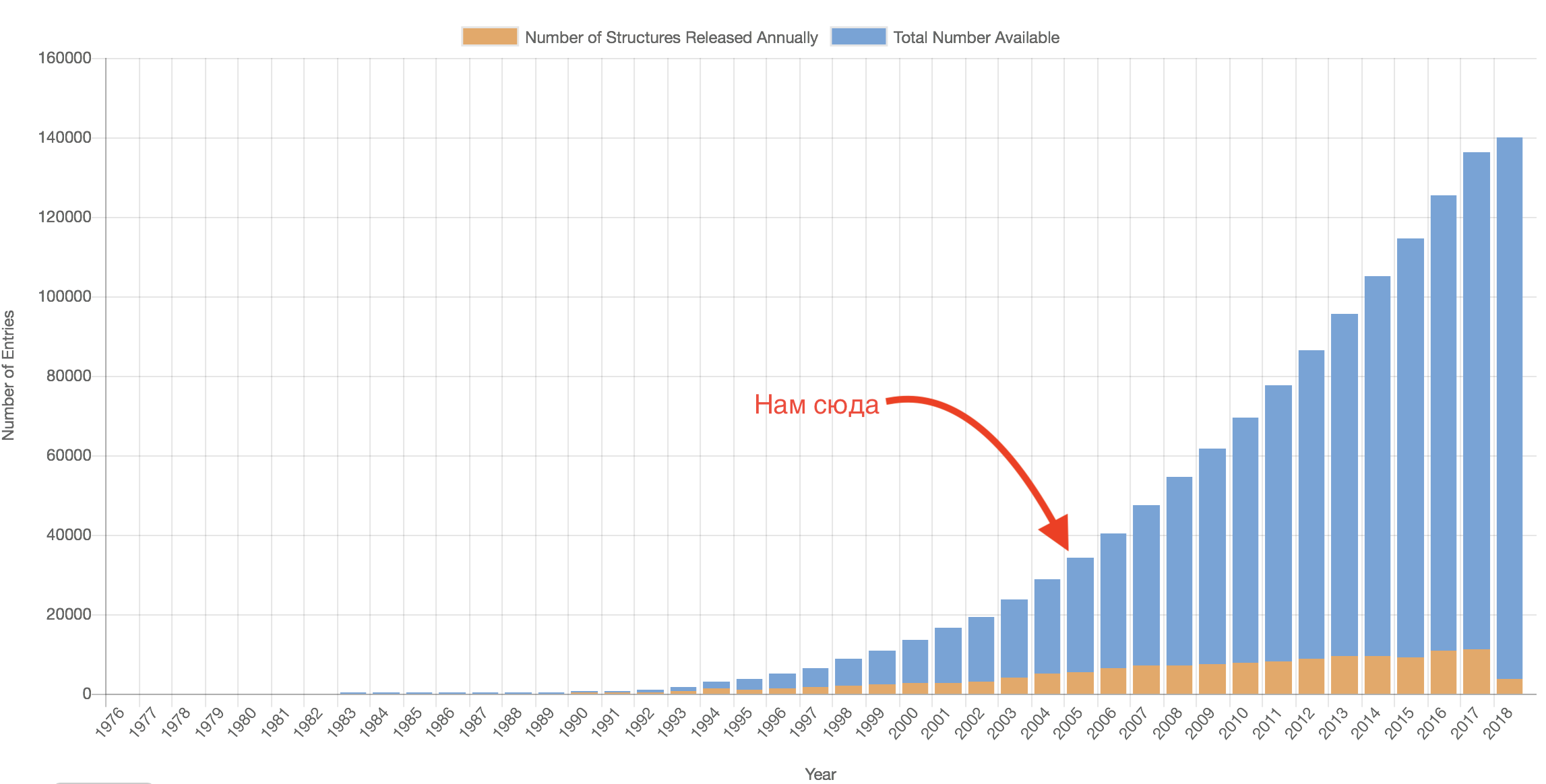
Итак, поехали. Предварительно нужно скачать весь Protein DataBank (у нас он был уже скачан ранее). Как это сделать, подробно можно прочитать здесь, там же дан удобный скрипт для скачивания всего добра через rsync. Мы же возьмем из него одну строчку, которая скачает нам только структуры по папочкам:
$> rsync -rlpt -v -z --delete --port=33444 rsync.wwpdb.org::ftp/data/structures/divided/pdb/ /path/to/your/local/dir > /tmp/pdb-download.log 2> /dev/null
Таким образом мы получаем все 130+ тысяч структур из RCSB PDB. Кажется, что это огромные данные, но мы будем работать с ними лишь примитивными инструментами командной строки, чтоб показать обратное. В первую очередь отберем все белки до 2005 года.
$> for filename in $(ls pdbs/*/*); do awk '$1 == "HEADER" {split($(NF-1),a,"-"); if (a[3] < 5 || a[3] > 18) {print FILENAME}}' $filename; done > pdbs_prior_2005.txt
На самом деле тот же код можно значительно ускорить, используя grep или ag для начальной фильтрации:
$> ag '^HEADER' pdbs | awk '{split($0,a,":"); split($(NF-1),b,"-"); if (b[3] < 5 || b[3] > 18) {print a[1]}}' > pdbs_prior_2005.txt
На маленьком множестве в 1000 файлов разница вот такая:
$> time (for filename in $(ls pdbs/*/*); do awk '$1 == "HEADER" {split($(NF-1),a,"-"); if (a[3] < 5 || a[3] > 18) {print FILENAME}}' $filename; done > /dev/null)
3,45s user 1,84s system 98% cpu 56,206 total
$> time (grep -e '^HEADER' pdbs/* | awk '{split($0,a,":"); split($(NF-1),b,"-"); if (b[3] < 5 || b[3] > 18) {print a[1]}}' > /dev/null)
7,34s user 0,19s system 100% cpu 7,530 total
$> time (ag '^HEADER' pdbs | awk '{split($0,a,":"); split($(NF-1),b,"-"); if (b[3] < 5 || b[3] > 18) {print a[1]}}' > /dev/null)
0,70s user 0,30s system 278% cpu 0,358 total
Очевидно, на выборке в 130+ тысяч стоит использовать ag 😉. Так или иначе, теперь у нас есть файл pdbs_prior_2005.txt, содержащий пути до всех файлов, чья дата загрузки в Protein DataBank раньше, чем 2005 год. Как и показано на графике выше, таких структур —
Теперь начнем отбирать по количеству альфа-спиралей. В первую очередь возьмем все белки с 30 и более спиралями. На этот раз сразу сделаем это эффективно:
$> cat pdbs_prior_2005.txt | xargs ag "^HELIX 30" | awk '{split($0,a,":"); print a[1]}' > pdbs_p2005_h30.txt
Следующая задача: найти 17 спиралей длиной не более 5 аминокислот. И вновь awk спасет нас, теперь благодаря возможности выполнения арифметических операций:
$> for filename in $(cat pdbs_p2005_h30.txt);do ag "^HELIX" $filename | awk 'BEGIN{i=0}{ if ($9-$6 < 6) {i++} }END{ if (i > 16) { print "'$filename'" } }'; done > pdbs_ph_h17.txt
Что же у нас получилось к текущему моменту?
$> wc -l pdbs_ph_h17.txt
2537 pdbs_ph_h17.txt
Размер файла уже достаточно разумный, всего 2537 записей, однако это всё еще слишком много, чтоб отсматривать глазами. Добавим правило про третью аминокислоту в альфа-спирали:
$> cat pdbs_ph_h17.txt | xargs ag "^HELIX 1" | awk '($6 < 3) { split($1,a,":"); print a[1] }' > pdbs_phh_aa3.txt
$> wc -l pdbs_phh_aa3.txt
142 pdbs_phh_aa3.txt
Теперь у нас есть всего 142 структуры!
Просмотрев их все глазами, мы смогли идентифицировать нужную — 1G72 (она была 117 в списке). Как оказалось, её изображение на сайте RCSB находится ровно в той же ориентации, что и на логотипе, а значит исходная идея с Хэммингом отлично бы сработала. Но это была замечательная возможность попрактиковаться в работе с инструментами командной строки над "большими" данными.