

Во всех околобиологических кругах мира эта неделя началась с обсуждения результатов CASP14, на котором вторая версия AlphaFold показала выдающиеся результаты в предсказании структур белков. Один только заголовок пресс-релиза DeepMind "AlphaFold: a solution to a 50-year-old grand challenge in biology" расколол общество так, что спустя несколько дней баталии в соцсетях всё еще продолжаются. Наверное, это повод сформулировать несколько мыслей на этот счет. Разумеется, без претензии на объективность.
Что произошло?
Раз в 2 года проходит соревнование Critical Assessment of Techniques for Protein Structure Prediction, известное так же как CASP. В ходе соревнования участникам предлагаются последовательности белков, структуры которых уже получены, но еще не опубликованы. Конкурсанты, имея только лишь эти последовательности, должны предсказать структуры этих белков. Задачи по предсказанию разделены на категории по сложности (в зависимости от того, насколько белок не похож на все уже существующие) и методам решения (полностью автоматизированные или с разной степенью человеческого участия).
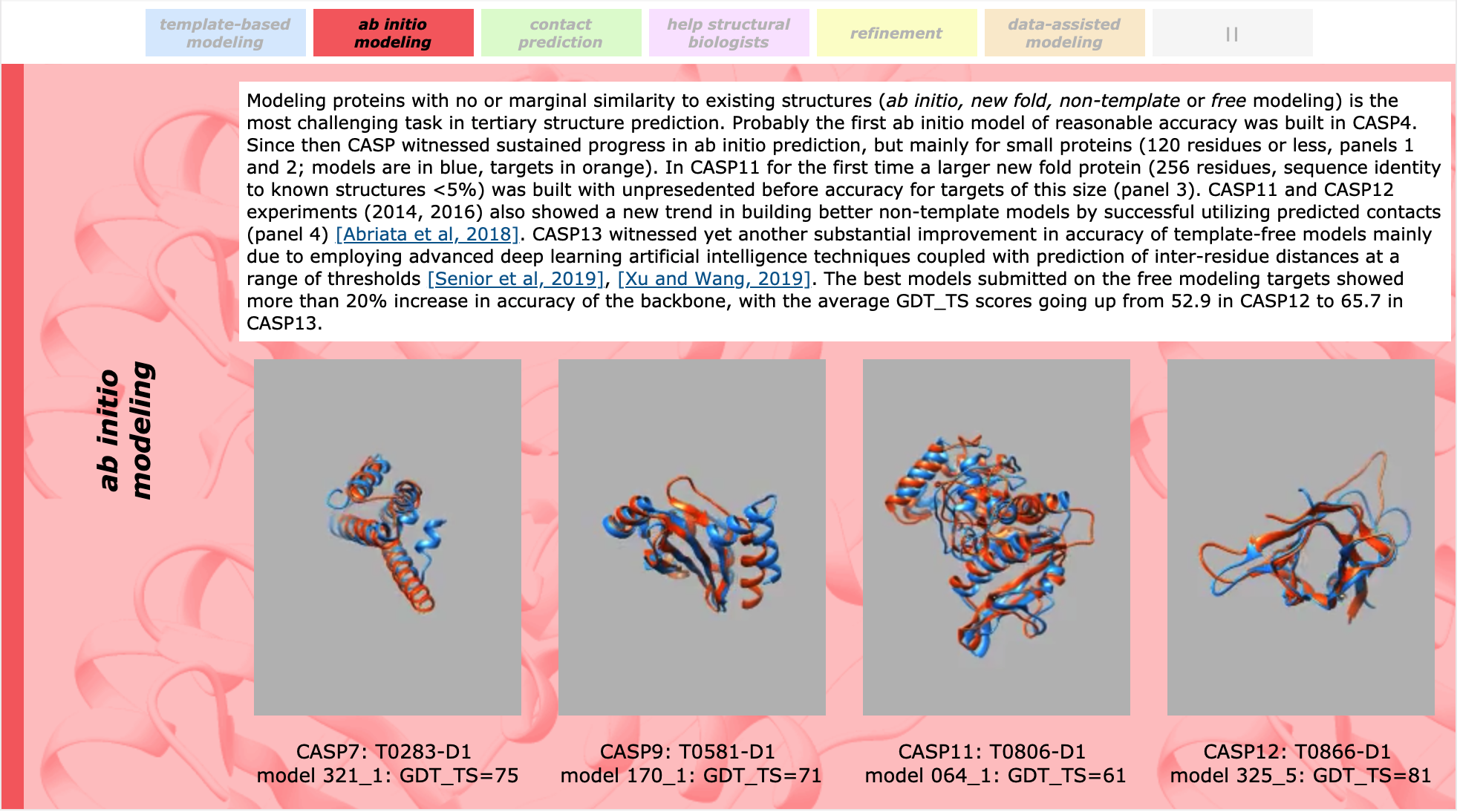
Традиционно самой сложной категорией считается ab initio modeling, то есть предсказание структуры "с нуля", без дополнительной информации о похожих белках. Долгое время прорывов в этой области практически не было, пока в 2018 году не появилась команда A7D, результаты которой на десятки попугаев (про них будет дальше) опережали все другие. Выяснилось, что под этим названием скрывался новый алгоритм AlphaFold от компании DeepMind, уже нашумевшей к тому времени победами на полях го. Разумеется, решение использовало глубокие нейросети, что вызвало огромный резонанс среди причастных к теме и, особенно, обывателей.
За прошедшие два года алгоритм был опубликован, код выложен, но революции в структурной биологии не произошло. В этом году результат был значительно улучшен: отрыв новой версии AlphaFold от второго места составил более 2.5 раз (в тех же попугаях), что подняло невероятную волну хайпа с одной стороны и негодования с другой. Прежде чем покритиковать мнения пессимистов и оптимистов, разберемся с постановкой задачи и интерпретацией результатов.
Про задачу
В первую очередь хочется определиться с терминологией. В своем пресс-релизе DeepMind использует выражение "protein folding problem". Формально, сворачивание (фолдинг) белка — это протяженный во времени процесс превращения цепочки аминокислот в пространственную структуру, несующую какую-либо биологическую функцию. От динамики и кинетики (по-простому, но не очень точно, траектории и скорости) этого процесса зависит как итоговая структура, так и, например, эффективность её производства, что представляет особый интерес для всех, кто занимается наработкой белков (например, фармацевтические компании).
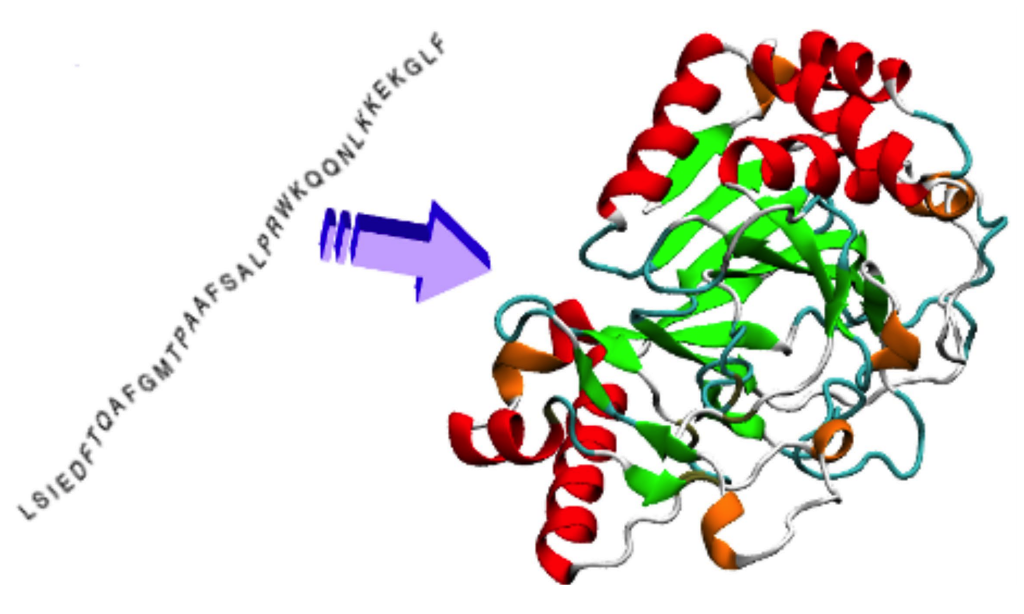
То, чем занимаются команды в ходе CASP, на самом деле имеет другое название — "protein structure prediction", то есть предсказание по аминокислотной последовательности только финальной структуры, без учета всего промежуточного процесса. Для огромного числа практических задач (можно даже аккуратно сказать "большинства") этого более чем достаточно. Более того, на сегодняшнем уровне развития только эта задача и является хоть сколько-нибудь решаемой (и то с большими оговорками), тогда как к моделированию процесса сворачивания мы подбираемся очень медленно. Симуляции для очень коротких белков (в видео ниже 39 аминокислот — в десятки раз меньше тех белков, с которыми обычно приходится работать) могут занимать несколько суток на распределенных GPU-кластерах. О получении правдоподобных траекторий для белков, представляющих реальный интерес (сотни аминокислот), пока не может идти и речи.
Как итог, большинство людей, как находящиеся в области, так и вне её, используют термин "фолдинг" как раз для задачи предсказания структуры. DeepMind немного смешивает понятия, говоря о решении "50-years-old grand challenge" в заголовке, но тут же поясняют, что они имеют ввиду именно предсказание структуры белков. Тем не менее, подобная небрежность позволила пуристам начать ворчать, что к проблеме фолдинга это всё не имеет никакого отношения. Разберем это чуть позднее, среди других претензий.
Даже редуцированная (относительно настоящего фолдинга) задача предсказания структуры белка все еще крайне сложна, поскольку требует определения положения каждого атома каждой аминокислоты. В реальных белках речь может идти о тысячах и десятках тысяч трехмерных координат, стремящихся обеспечить минимальность энергетического потенциала. Как выглядит функция, приближающая значение потенциальной энергии такой системы, мы более-менее представляем. Но вот размерность задачи напрочь убивает возможность применения подходов, связанных с глобальной оптимизацией, а локальная не подходит из-за, мягко говоря, невыпуклости, потенциала. Если последние несколько предложений непонятны, то ключевая мысль такова: хоть мы и знаем как найти решение "в лоб", никакой компьютер ни за какое (разумное) время не сможет этого сделать. Из-за этого приходится идти на разные ухищрения. Будем их рассматривать на примере одного коротенького белка из 28 аминокислот.
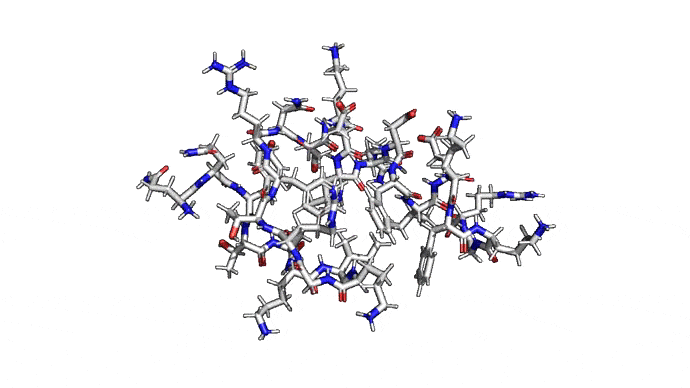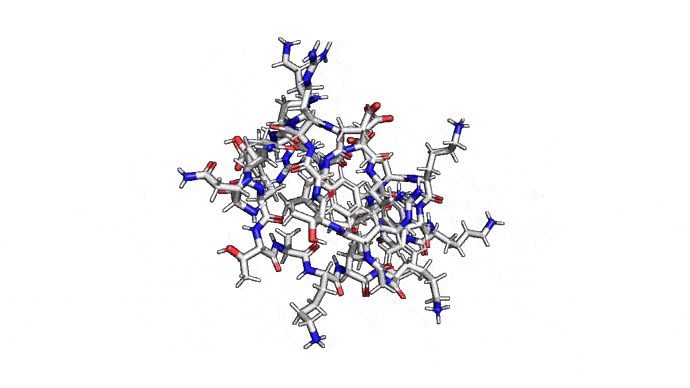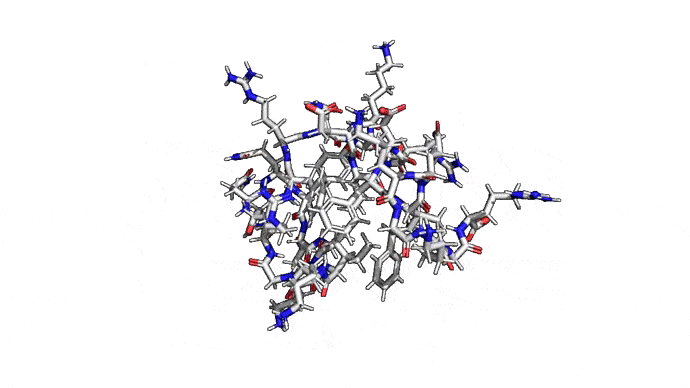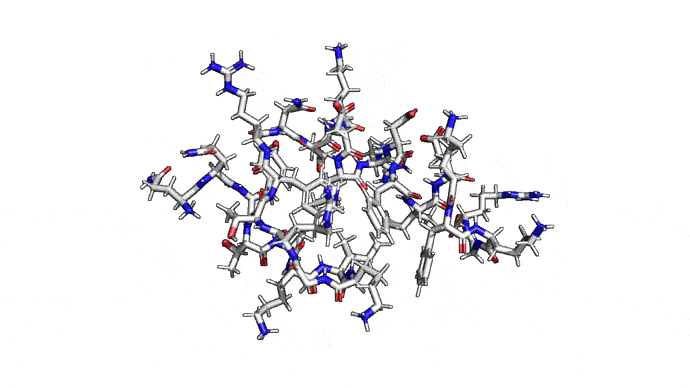
Самым распространенным подходом к предсказанию структуры является предсказание по шаблону (template-based), он же гомологичный фолдинг. В его основе лежит идея о том, что похожие последовательности аминокислот будут давать похожие пространственные структуры в белках. Имея достаточно большую базу качественных экспериментальных структур можно, заменяя ряд фрагментов, получить довольно качественную полноатомную модель. Дополнительные знания о конкретном классе белков позволяют заполнять отличающиеся или неизвестные фрагменты особенно хорошо. То есть гомологичный фолдинг — это такое отражение идеи, что если задачу нельзя решить для всех белков, то её можно решить по крайней мере для каждого. Не универсально, зато работает.
Проблемы, очевидно, начинаются, когда близких по последовательности белков в базе структур не находится, что, конечно, случается довольно часто. Эта задача считается столь сложной, что её урезают еще сильнее. Но продолжают называть так же, да. Это биология.
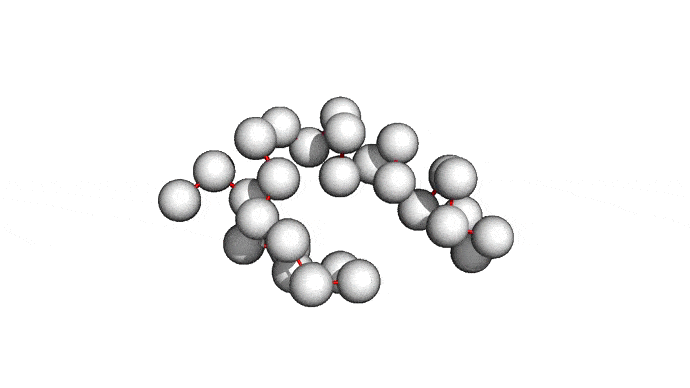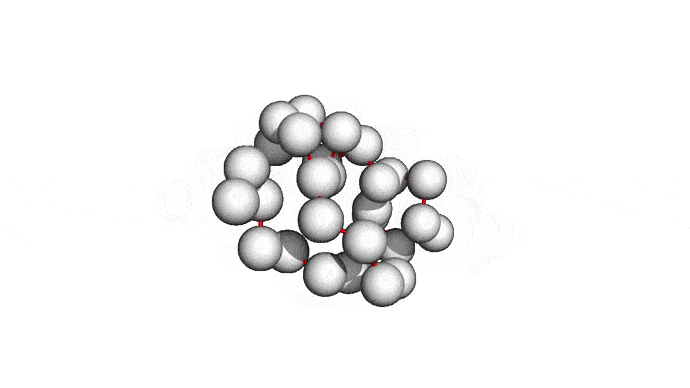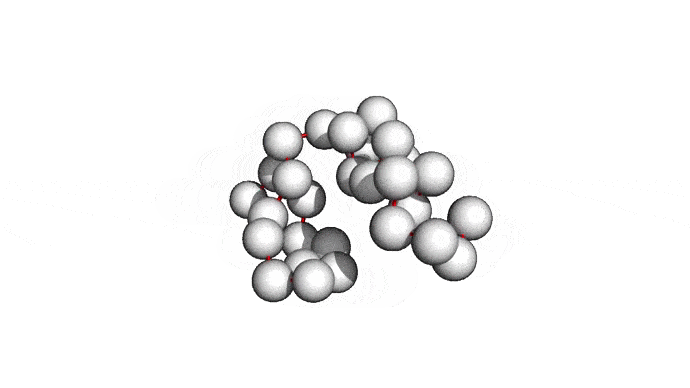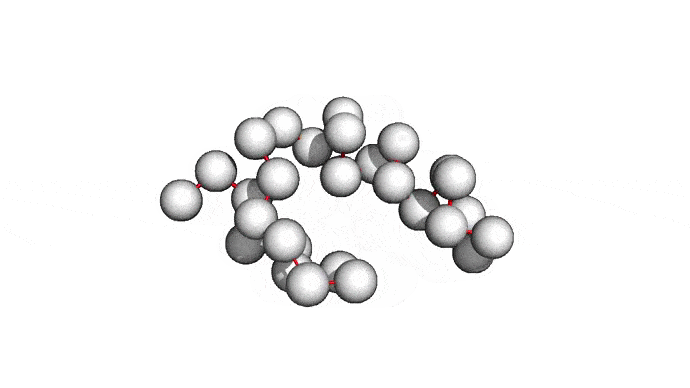
Как можно сократить задачку? Например, выкинуть большую часть атомов. В первую очередь (уже подвох!) откажемся от атомов радикалов, то есть обезличим каждую аминокислоту, оставив только атомы основного каркаса. Для многих задач такая структура уже не подойдет, но мы все еще отлично сможем понимать общее устройство белка. А забегая вперед, скажу, что мы умеем довольно неплохо восстанавливать положения радикалов, если структура каркаса у нас уже есть. Вот что у нас получится:
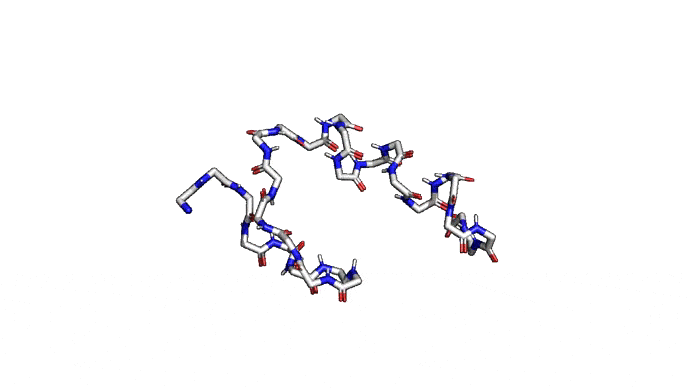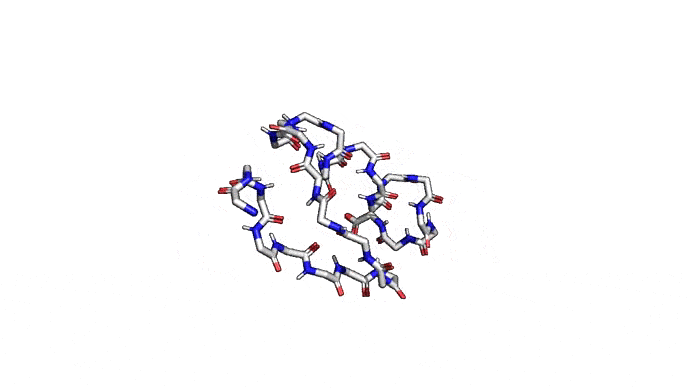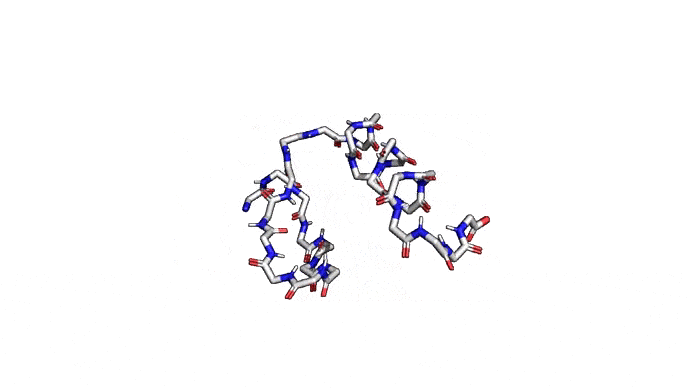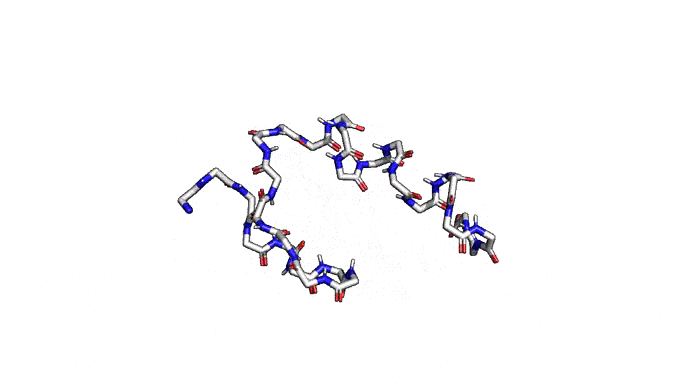
Можно пойти еще дальше, а именно представлять каждую структуру и вовсе одним атомом \(C_{\alpha}\) — углерода, из которого "растет" радикал аминокислоты. Такая модель будет совсем грубой, но зато количество атомов сократится совсем значительно и будет равно количеству аминокислот. Использовать такую структуру для решения каких-либо прикладных задач, конечно, будет практически невозможно. Но, авось, потом дотянем. Именно это и предлагается делать в категории ab initio modeling соревновании CASP.
Про попугаев
Мы запихнули нашу последовательность аминокислот в предсказатель и на выходе получили набор координат для наших \(C_{\alpha}\) атомов. Как теперь понять, хороши мы или нет? Для этого требуется выровнять молекулы, то есть расположить их так, чтоб соответствующие друг другу атомы оказались как можно ближе друг к другу, а затем посчитать расстояния между соответствующими парами атомов. Одно такое расстояние между атомами двух моделей одной структуры показано ниже:
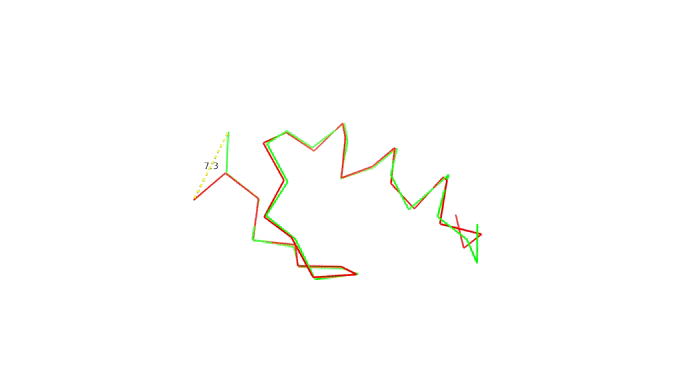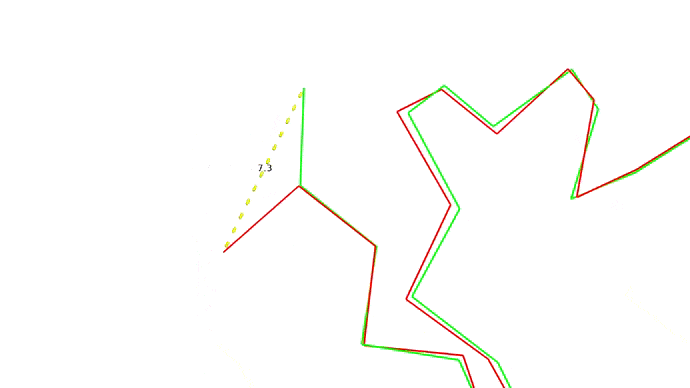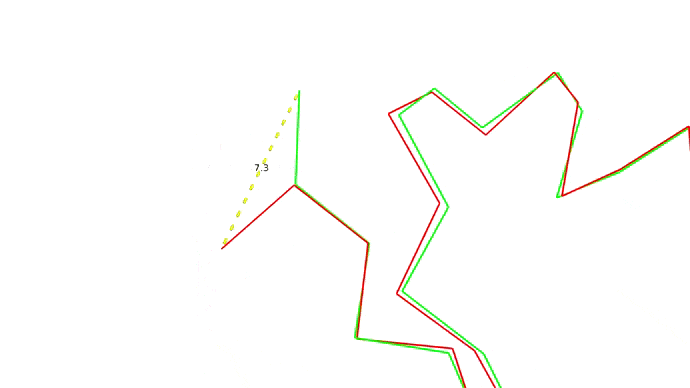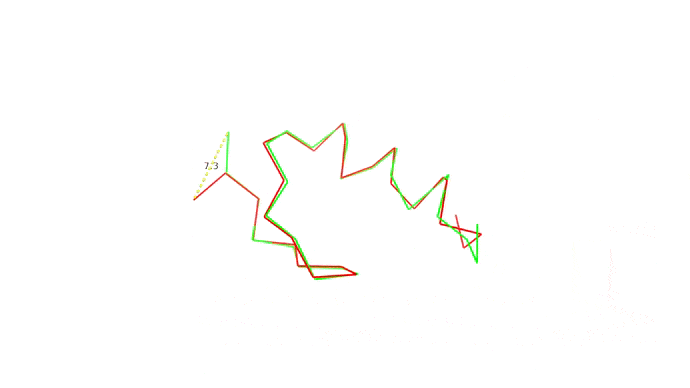
Найдя расстояния между каждой парой атомов мы можем оценить близость структур друг к другу всего одной цифрой RMSD — root-mean-square deviation, то есть среднеквадратичное отклонение атомов друг от друга. RMSD между моделью и "истинной" (то есть полученной экспериментально) структурой и есть оценка качества моделирования.
\[ RMSD_{s_1, s_2} = \sqrt{\frac{1}{num\_atoms(s_1)}\sum\limits_{i \in num\_atoms(s_1)} |s^{i}_1 - s^{i}_2|^2} \]
Очевидной проблемой RMSD является усреднение. То есть предсказания, в котором все атомы предсказаны правильно, а один очень далеко (как на картинке выше), и предсказание, в котором все атомы предсказаны не очень, могут получить одинаковую оценку, хотя с практической точки зрения они имеют очень разное качество. В связи с этим CASP использует оценку на основе Global Distance Test (GDT). Идея GDT состоит в подсчете доли \(C_{\alpha}\) атомов модели, которые после выравнивания оказываются на расстоянии не более некоторой отсечки от соответствующих им \(C_{\alpha}\) атомов "истинной" структуры. Таким образом GDT распределена от 0 до 1, и обычно измеряется в процентах. В случае CASP используют отсечки в 1, 2, 4 и 8 Å, после чего усредняют полученные 4 показателя и выводят одну цифру, называемую "total score" — GDT_TS.
\[ GDT\_TS = \frac{1}{4}(GDT_{1Å} + GDT_{2Å} + GDT_{4Å} + GDT_{8Å}) \]
На самом деле, организаторы CASP идут еще дальше, насилуя даже полученные показатели GDT_TS путем превращения их в z-score (по выборке всех предсказаний данной структуры), выкидывая выбросы и обрезая слишком низкие значения (-2 или 0). Z-score по всем предсказанным структурам для данной команды дальше и суммируются и усредняются. В итоге мы получаем 4 показателя: суммарные z-score для отсечек -2/0 и средние z-score для отсечек -2/0. Обычно это делается для того, чтоб уйти от необходимости объявлять победителя, оставив возможность сразу нескольким командам сказать, что они №1(2,3...) в соревновании CASP по такому-то показателю.
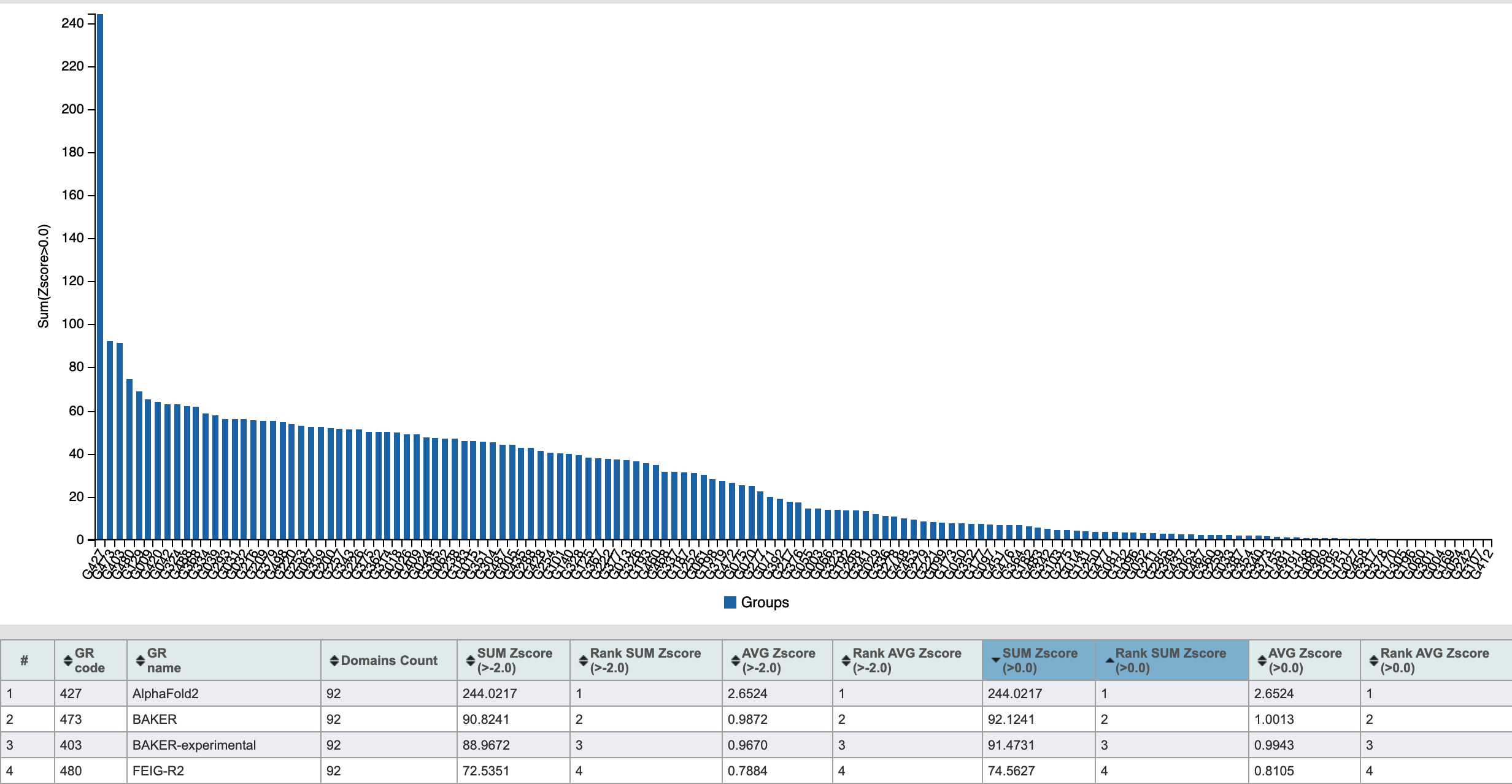
В этот раз по всем четырем показателям безоговорочно выиграл AlphaFold2.
В своем пресс-релизе DeepMind также приводит медианные значения GDT_TS для всех мишеней — 92.4, а также для особо сложных белков — 87.0. Первый показатель, по их прикидкам, соответствует RMSD в 1.6 Å, что, формально, сопоставимо с качеством структур, получаемых в ходе рентгеноструктурного анализа. Однако это заявление, как и многие другие, несколько спекулятивно.
Как эта штука работает?
Мы достоверно не знаем, как именно работает AlphaFold2, но можем предполагать на основе пресс-релиза, опубликованной статьи и кода первой версии алгоритма. На данном этапе это, наверное, даже хорошо, поскольку не приходится терзаться сомнениями о том, насколько подробно рассказывать про его устройство.
Вместо этого посмотрим на основные идеи, благодаря которым вся затея выстрелила. По моему мнению, она вовсе не в хитрых архитектурах нейросетей. Они, конечно, будут всё сложнее, что поможет выжимать всё больше и больше попугаев в соревнованиях. Но всё же главным залогом успеха стало применение сверточных нейросетей в совокупности с полезным наблюдением (сделанным не DeepMind), которое позволило грамотно готовить данные и выбрать формат выхода предсказателя. С последнего и начнем, но прежде на всякий случай еще раз повторюсь: мы рассмотрим пару общих идей. Некоторые вещи, специфичные для AlphaFold, я умышленно опускаю.
AlphaFold не предсказывает структуру белка.
Давайте на секунду остановимся и задумаемся над тем, а как вообще математически можно выразить структуру белка. До того мы постоянно неявно подразумевали, что это координаты атомов (всех, каркасных, \(C_{\alpha}\) или каких-то ещё), но на практике это очень неудачное представление, поскольку оно не единственное. Мы обычно считаем, что предсказание работает как некоторая детерминированная функция: принимает на вход последовательность и всегда возвращает один единственный ответ. Но какой из бесконечного набора координат "канонический"?
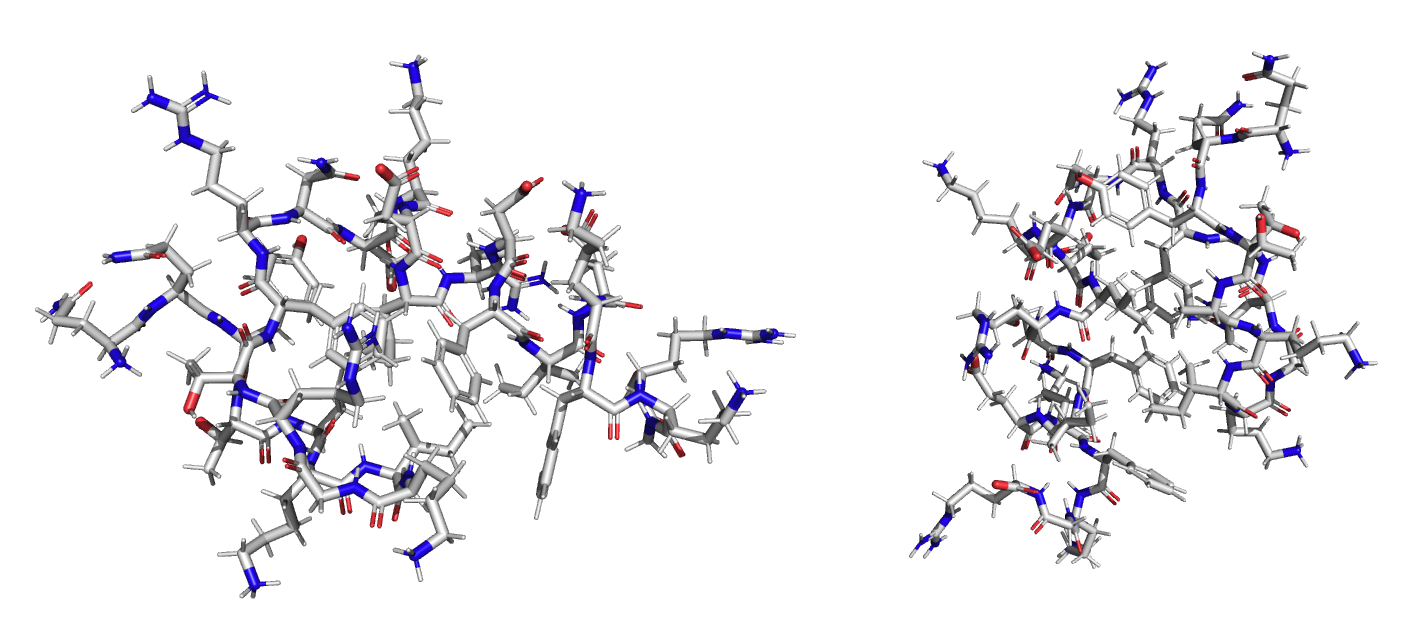
Дополнительно совершенно неясно, как на этом тренировать модели машинного обучения. Ведь они должны искать закономерности в данных, а если все белки будут по-разному повернуты и перенесены в пространстве, то находить что-либо в совершенно ничего не значащих координатах занятие заведомо бесперспективное.
Поняли это, конечно, не DeepMind, так что поиском инварианта, то есть какого-то представления структуры, не зависящего от её поворотов и переносов в пространстве, занялись довольно-таки давно. Классическим решением последних десятилетий был подход, основанный на углах.
Проведем простой эксперимент на всё той же маленькой структуре, с которой мы работаем всю дорогу. Измерим расстояния между всеми его последовательно идущими \(C_{\alpha}\) атомами.

Внезапно оказывается, что эти расстояния практически равны. Более того, даже если мы возьмем другой белок и посмотрим расстояния в нем, мы увидим ровно те же значения. Я не поленился и сделал это для нескольких последовательно идущих \(C_{\alpha}\) атомов структуры белка NTL9 из видео в начале заметки:

Что ж, это весьма значительное наблюдение позволяет нам предположить, что мы можем достраивать нашу структуру из \(C_{\alpha}\) атомов только по направлениям следующего атома относительно предыдущего, используя при этом постоянные расстояния.
Если бы мы жили в координатном пространстве, то такое направление задавал бы какой-нибудь единичный вектор. Но мы помним, что координатное пространство нам не нравится тем, что оно абсолютно, а наши белки могут двигаться в этом пространстве как угодно. Было бы здорово указывать это направление тоже как-то относительно, без привязки к глобальному пространству. Для этого нам и потребуются углы.
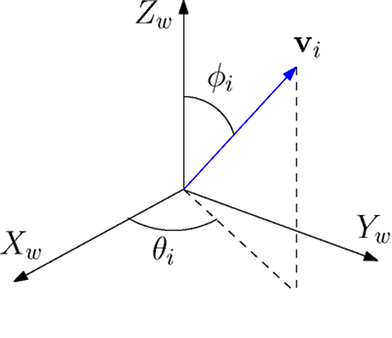
Картинка выше должна убедительно показать, что направление вектора в трехмерном пространстве может быть легко задано двумя углами (можем считать их широтой и долготой, как на глобусе). Однако чтоб задать эти углы нам нужна некоторая плоскость, относительно которой они будут заданы и точка на плоскости, относительно которой мы будем задавать направление.
С точкой всё ясно — это просто предыдущий атом. Плоскость, как известно, может быть задана тремя точками. Мы можем взять три предыдущих атома, и тогда два угла возникают очень естественно. Первый считается как плоский угол между тремя атомами (текущим и двумя предыдущими). Второй — как двугранный угол по всем четырем точкам.
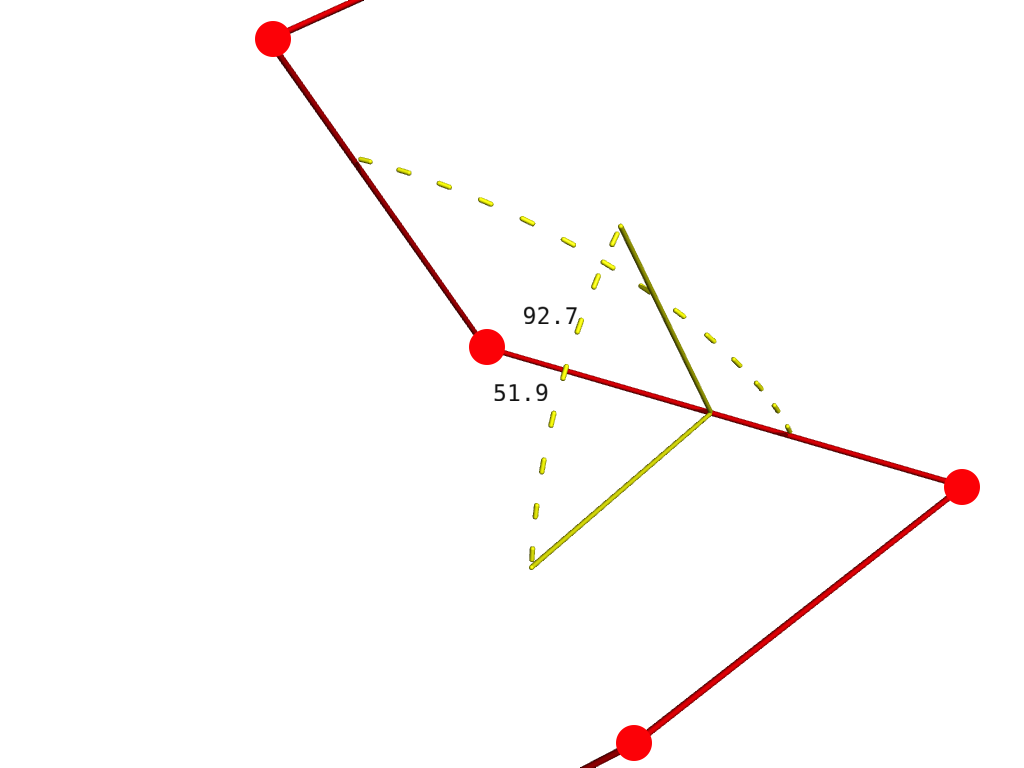
Представление довольно-таки удобное, так как позволяет для N аминокислот предсказывать N-3 двугранных и N-2 плоских углов, которые полностью определят структуру. С ним есть только одна проблема, оно не работает.
Если быть точнее, никто так и не заставил какие-либо модели прилично предсказывать углы по последовательности. Конечно, таких алгоритмов великое множество, но все они работают с разной степенью паршивости. Почему так происходит, пожалуй, тема отдельной заметки, но в двух словах, кажется, что это связано с тем, что предсказание углов — это решение задачи про очень локальную область структуры, не учитывающее никаких взаимодействий аминокислот, находящихся далеко друг от друга.
И вот здесь на сцене появляется другой, гораздо более простой инвариант. В отличии от углов его можно объяснить буквально одной фразой — инвариантом структуры является матрица (таблица) всех попарных расстояний между атомами. Такое представление не зависит от поворотов или перемещений структуры в трехмерном пространстве, содержит информацию о взаимодействии (близости) аминокислот вне зависимости от их удаленности по последовательности. Также человечество уже очень давно умеет эффективно находить набор координат по матрице расстояний. Это тоже тема для отдельной заметки, но самостоятельно посмотреть один из старейших методов можно, например, здесь.
0.0 3.83271 6.01989 6.58519 … 25.678 24.6388 24.0295
3.83271 0.0 3.83997 6.59813 23.5736 22.5554 21.486
6.01989 3.83997 0.0 3.82077 20.5022 19.164 18.2255
6.58519 6.59813 3.82077 0.0 20.6506 19.0817 18.7819
10.0775 9.08123 5.37992 3.8444 17.0151 15.2753 15.0675
11.085 10.8644 7.81588 5.66081 … 16.316 15.1335 15.7933
14.4674 13.5387 10.1526 8.7795 12.6211 11.3417 12.3157
16.4536 15.7524 12.8618 11.6161 11.6552 11.4539 13.233
13.8395 13.8638 11.4989 9.82352 15.386 15.1816 16.6443
10.8774 10.5668 8.77125 8.06832 16.6945 16.5649 17.2367
7.27608 6.78375 5.36449 5.38269 … 18.9654 18.3924 18.3666
8.49109 6.13378 4.79525 7.04693 17.8971 17.399 16.6753
8.15412 4.97326 5.92916 9.24726 20.7562 20.3603 19.1268
⋮ ⋱ ⋮
14.6443 11.3347 9.85678 12.5927 14.7822 14.6226 12.817
13.4068 10.4863 7.84574 9.82696 13.6534 12.7312 11.2792
16.7364 13.568 10.9384 12.9258 12.5891 11.3365 8.88843
18.6057 15.4437 13.2634 15.3405 11.0792 11.0058 8.80482
17.6728 15.0817 12.5043 13.7692 … 9.06338 9.17429 8.31031
19.7891 17.344 14.2232 14.8957 6.80036 5.72403 4.80765
22.4295 19.6197 16.8814 18.1163 6.56397 6.69385 4.6497
22.6069 20.0842 17.5949 18.6529 5.71467 7.68905 7.47085
22.0187 19.9578 17.0287 17.2996 3.8602 5.29157 6.763
25.678 23.5736 20.5022 20.6506 … 0.0 3.81372 6.17142
24.6388 22.5554 19.164 19.0817 3.81372 0.0 3.84365
24.0295 21.486 18.2255 18.7819 6.17142 3.84365 0.0
Именно эту матрицу и ищет AlphaFold. Поиск же координат происходит уже существующими инструментами с помощью градиентного спуска. Также AlphaFold предсказывает расстояния не только между \(C_{\alpha}\), но также берет в рассмотрение и \(C_{\beta}\) — первые атомы радикалов аминокислот.
Теперь, когда нам стало понятно что предсказывает AlphaFold, и как это превращается в структуру, сделаем еще одно громкое заявление.
AlphaFold получает на вход не последовательность аминокислот белка.
К слову, именно это является главной проблемой в использовании выложенного DeepMind кода (но есть вариант имплементации от сообщества, исправляющий эту ситуацию). Для того, чтоб получить структуру интересующей последовательности её нужно правильно подготовить. Но именно в этой подготовки таится вторая важная составляющая успеха.
Сама по себе последовательность, очевидно, не несет никакой информации о взаимодействии аминокислот, в связи с чем какой бы хитрой и сложной ни была нейросеть, извлечь эту информацию ей довольно-таки тяжело. Мы можем упростить эту задачу, если заранее сообщим нашему алгоритму, что какие-то позиции в нашей последовательности, возможно, имеют связь друг с другом. Но откуда эту информацию взять?
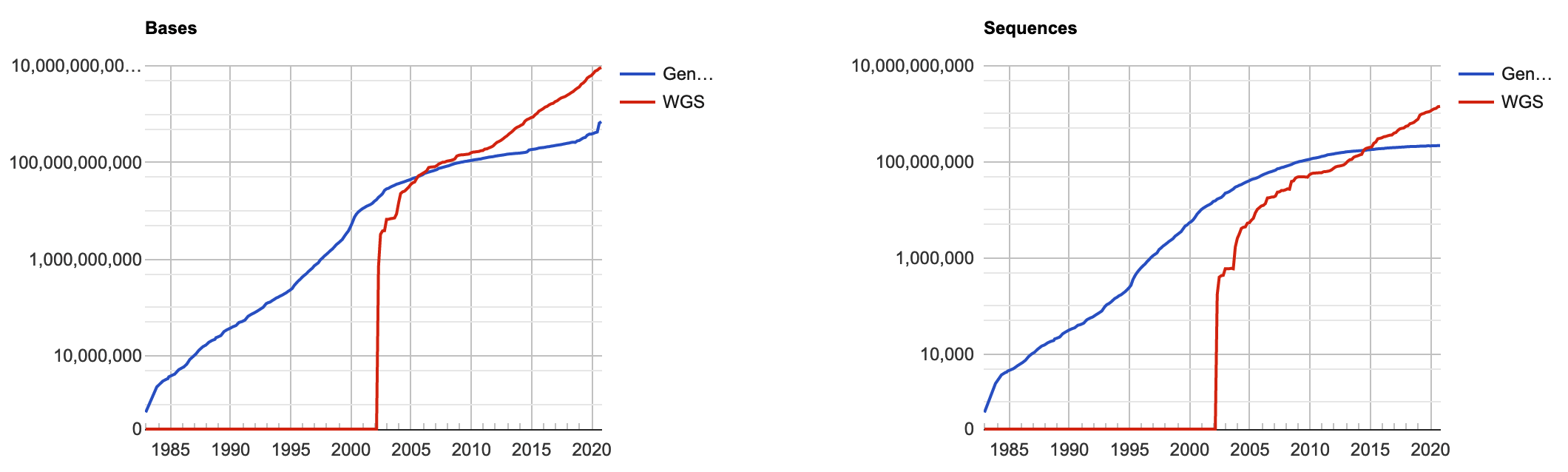
Даже то, что для нашего белка отсутсвует какая-либо структурная информация не означает, что мы не знаем про него ничего. С момента изобретения методов высокопроизводительного секвенирования люди добыли и разметили поистине колоссальное количество последовательностей генов, кодирующих разные белки. И среди них обязательно найдутся последовательности, хотя бы отчасти похожие на интересующую нас.
Скорее всего эти последовательности похожи не просто так, а как раз потому, что кодируемые ими белки выполняют схожие функции (в том же или другом организме). Это, очевидно, означает, что и структура этих белков должна быть похожа.
"Ну, и что?" спросите вы. Ведь мы не знаем структуры этих последовательностей. Зачастую мы даже их названий не знаем. Чем нам может помочь, что кто-то в каком-то организме увидел похожую последовательность? На самом деле, очень многое.
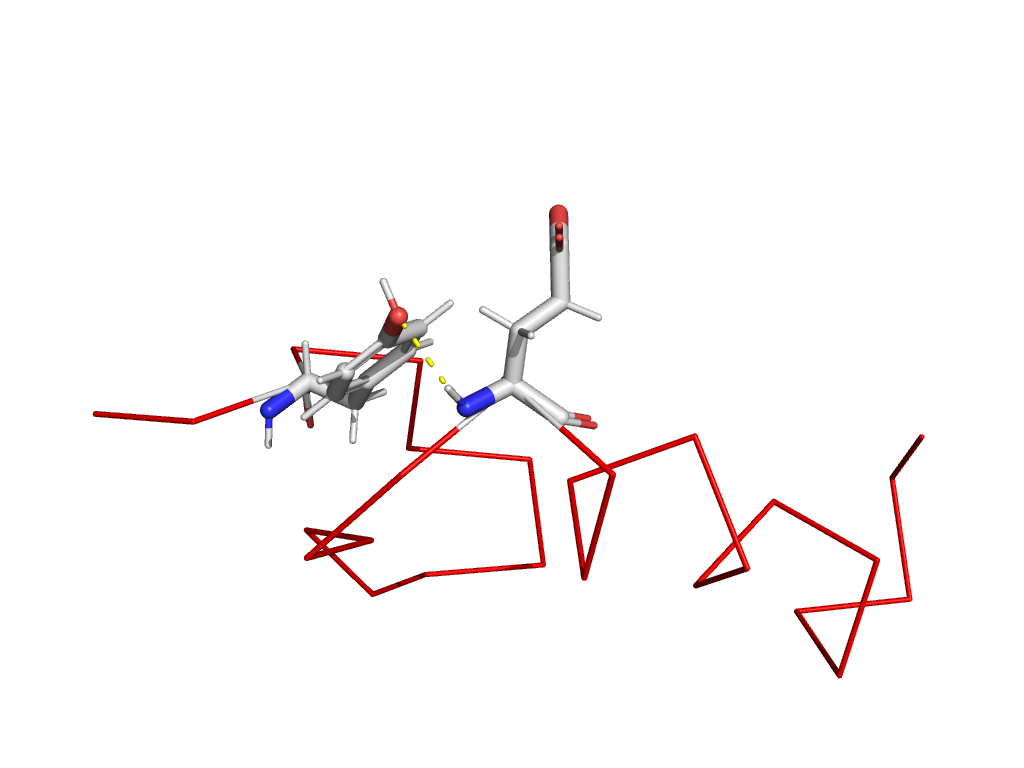
Давайте вновь посмотрим на наш белок и сосредоточимся на паре взаимодействующих аминокислот. Давайте представим, что мы заменим одну из них. В этом случае взаимодействие с высокой вероятностью будет утрачено, белок изменит свою структуру и, возможно, даже перестанет выполнять свою функцию. Организм в котором произойдет такая мутация, скорее всего, пострадает и с меньшей вероятностью даст потомство.
С другой стороны, если эта замена произойдет на какую-то аминокислоту, способную образовать такую же или похожую связь, то структура поменяется несильно и функция, вероятно, утрачена не будет.
Это наблюдение имеет самое прямое отношение к нашим последовательностям. Мы наблюдаем результаты секвенирования из жизнеспособных организмов. Из этого мы делаем выводы, что гены в них, скорее, работают как надо. А значит, структуры схожих последовательностей будут иметь схожие взаимодействия. Позиции, обеспечивающие эти взаимодействия, скорее всего будут скоррелированы, то есть меняться совместно. Посчитать такую корреляцию по корпусу найденных похожих на нашу последовательностей довольно просто.
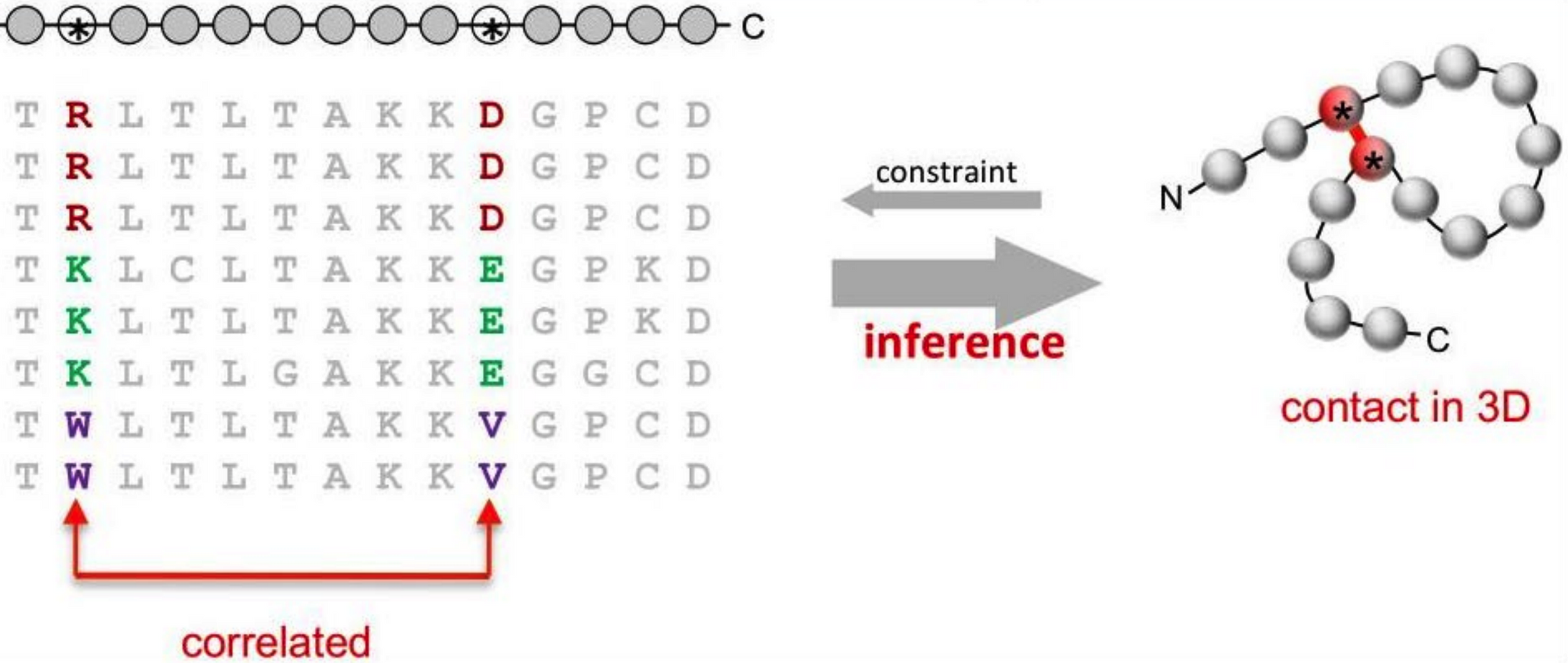
Так, помимо последовательности, которая сама по себе несет очень мало информации, мы еще получим квадратную матрицу, описывающую корреляцию каждой пары позиций (её обычно называют матрицей коэволюции). Кстати, она будет такого же размера, что и ответ алгоритма — матрица попарных расстояний (тоже NxN при длине последовательности N). Это приятно, поскольку значительно упрощает построение сверточной нейросети, которая будет с этим делом работать.
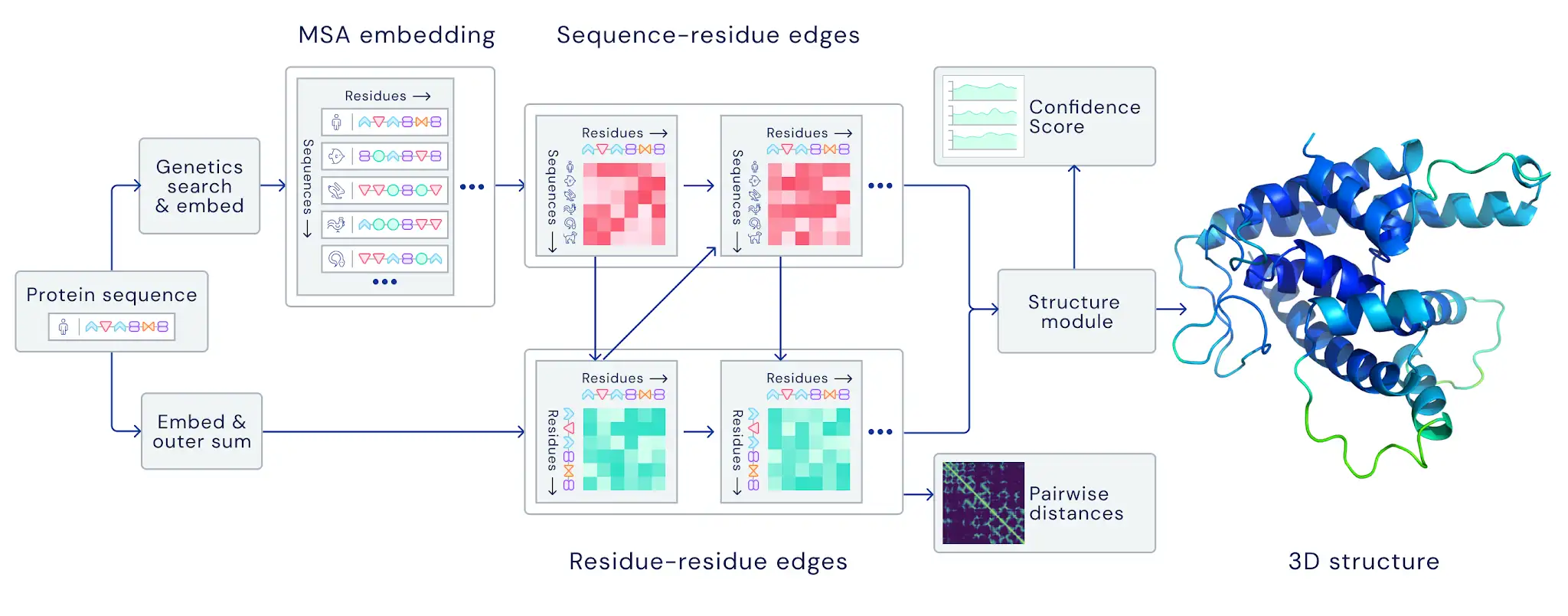
В пресс-релизе AlphaFold2 очень вскользь сказано, что новая нейросеть была обучена end-to-end. Из этого можно осторожно предположить, что нейросеть теперь берет на себя все этапы от поиска коэволюции до финального представления структуры. Правда там же на изображении показан этап поиска близких последовательностей (Genetics search & embed), который, по всей видимости, делается всё же более классическими методами. Так или иначе, судить об этом только лишь по PR-новости рано, будем ждать статью.
Если вы дочитали аж досюда, то уже догадались, что я вас обманул и под видом комментария к религиозным спорам в соцсетях выдал текст о задаче предсказания структуры белков и идей, позволяющих эту задачу решить. Честно говоря, проследить эволюционный процесс приведший нас к результатам CASP14, мне действительно интереснее, чем обсуждать частные мнения относительно применимости этих результатов. Но давайте всё-таки посвятим им несколько абзацев в блиц-режиме.
Что говорят пессимисты
Это же не фолдинг!
Я несколько раз встречал высказывания, что DeepMind всех обманывает, выдавая одну проблему за другую. Ведь решалась задача предсказания структуры белка, а не процесса фолдинга. А значит, говорят такие люди, и ценности тут нет никакой.
В процессе повествования я уже отмечал, что применение термина "folding problem" к задаче предсказания структуры повсеместен, так что таких критиков я записываю в разряд тех самых пуристов, что, на самом деле, неплохо. Но важной роли эти высказывания, на мой взгляд, не играют, во всяком случае, пока мы верим в то, что аминокислотной последовательности достаточно для того, чтоб предсказать структуру. По большей части, мы в это верим, но есть нюансы, о которых будет немного позже.
А вот соображения, что практическая ценность предсказания структур низка мне кажутся довольно вредными передергиваниями. Действительно, для решения ряда важнейших проблем было бы крайне полезно знать путь, по которому белок сворачивается к итоговой форме, но подавляющее большинство практических задач требует именно качественных структур (но, конечно, не исчерпывается ими). Возможность когда-нибудь не тратить по 2 года на получение таких структур экспериментальными способами была бы настоящим прорывом.
Это работает только с однодоменными белками
Многие белки упакованы не единым доменом, а состоят из нескольких последовательно упакованных фрагменов — доменов. Типичным примером являются столь близкие мне антитела.
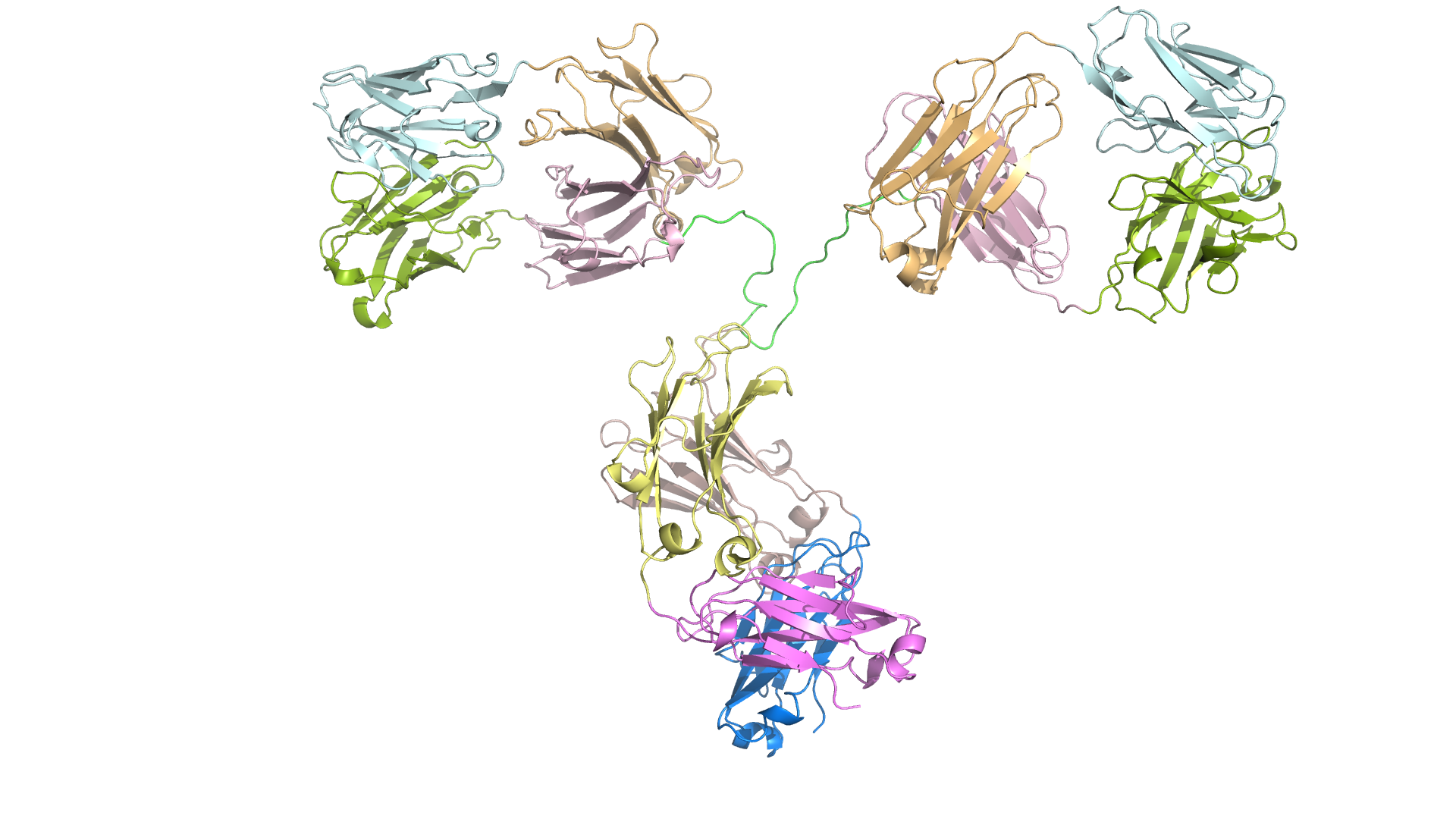
Ограничение довольно серьезное, но не непреодолимое. Мне кажется, принципиальных ограничений, которые бы препятствовали получению структуры многодоменных белков нет. Но текущее решение, кажется, пока решать такие задачи не позволяет.
Гораздо интереснее, когда будут методы, позволяющие получать структуры белков, состоящих из нескольких независимых цепей аминокислот. Публикации по всё тем же антителам намекают, что и здесь особых проблем нет, и модифицировать нейросеть для приема сразу нескольких цепей вполне возможно. К тому же образование таких белков ближе к задаче поиска состыковки молекул (молекулярный докинг), а потому, формально, может вообще не рассматриваться в контексте предсказания структуры. Но это вопрос вкусовых предпочтений.
Предсказываются усредненные структуры
На самом деле, соображение о том, что аминокислотная последовательность однозначно определяет структуру абсолютно ложно. Ничуть не меньше на это влияют условия, в которых сворачивается белок: концентрация различных ионов, присутствие поблизости других белков и так далее. В зависимости от этих условий результирующие структуры могут быть совсем различны.
Тренировка AlphaFold (как и всех прочих алгоритмов машинного обучения для решения данной задачи) происходит на базе экспериментальных структур Protein Data Bank (PDB), которая содержит белки в самых различных условиях. Там запросто можно найти множество белков с одинаковой последовательностью, но различными структурами.
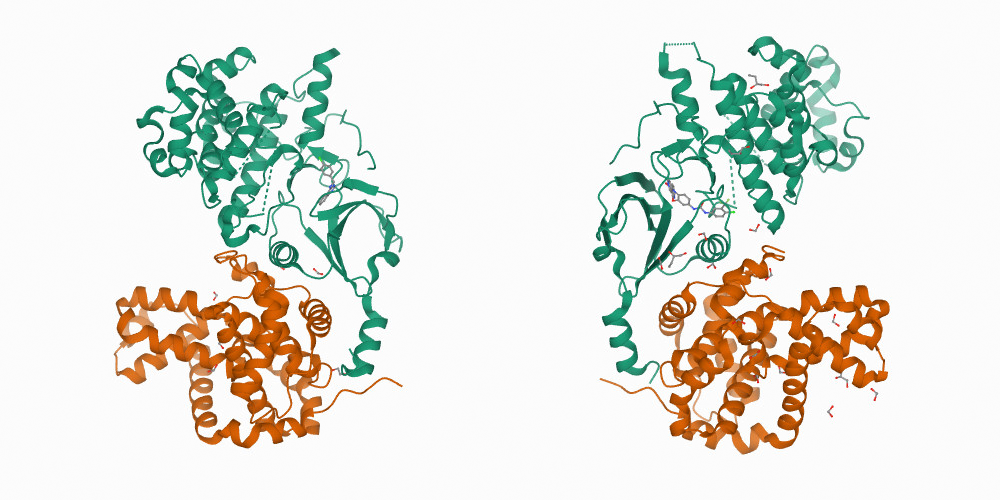
Для нас это означает, что AlphaFold усредняет структуры, получая результаты, которые могут не соответствовать никаким реальным условиям, а, значит, такие результаты бесполезны.
На практике, всё, конечно, сложнее. Действительно целые классы белков имеет смысл рассматривать только вместе с условиями, поскольку их механизм действия именно в том, что они по-разному функционируют в разных ситуациях. Но таких объектов явно не большинство.
При разработке лекарственных средств мы, преимущественно, сталкиваемся с объектами, для которых условия не оказывают значительного влияния. Все дизайны антител к поверхностным рецепторам, молекул к киназам и капсидов вирусов для генотерапии в подавляющем большинстве случаев делаются обычно без учета условий, как раз для усредненного случая. В ситуациях, когда они всё же нужны, мы прибегаем к молекулярным симуляциям, где наш белок уже можно "посолить" нужным образом. Всё-таки предсказание структур — это не единственный метод структурной биоинформатики, и нужно просто грамотно применять другие.
Однако надо честно признать, что есть задачи, в которых это всё не сработает. Изменения структуры в различных условиях столь значительны, что вычислительные методы пока не способны их симулировать в динамике. В этом случае нам нужны структуры, полученные в интересующих условиях. Проблема в том, что если мы начнем делить обучающую выборку на подвыборки в заданных условиях, 170 тысяч структур из PDB может уже и не хватить, чтоб нормально обучить модель. А, может, и хватит. В любом случае, AlphaFold сейчас так действительно не умеет.
Мы не знаем границ применимости метода
В случаях, когда мы предсказываем структуры неизвестных белков из новых семейств, мы понятия не имеем, как выглядит ответ, а потому не можем проверить истинность предсказания. Таким образом мы получаем просто некоторый ответ, не зная, когда он верный, а когда нет.
Не то, чтоб эта ситуация была нетипичной для глубокого обучения — обычно при решении задач нейросетями мы знаем и того меньше. Сможете ли вы проинтерпретировать и понять, почему тот или иной ответ выдала GPT-3 с миллионами параметров? Да, ни в жизни. Тем не менее это не мешает людям её применять. Точно так же мы используем абсолютно неинтрепретируемые модели в компьютерном зрении, торговле на бирже, самоуправляемых автомобилях и многом другом. Так что обычно это не является проблемой.
Сообщество структурных биоинформатиков (да, и вообще людей, работающих в биотехе) в этом вопросе более щепетильны и не любят использовать результаты, которые не могут оценить на адекватность. И это, конечно, правильно. Но, боюсь, слепое доверие машинному обучению однажды накроет и эту гавань.
Точности всё еще не хватает для рационального дизайна
В качестве аргумента на эту точку зрения приводятся ситуации, когда изменение положения атомов на 1 Å приводит к значительному изменению функции белка. Я таким людям хочу задать вопрос: а много вы видели экспериментально полученных структур с разрешением лучше 1 Å? Чтоб никого не утруждать, сам же на него и отвечу. Таких структур 803 штуки. Из 170 тысяч, да.
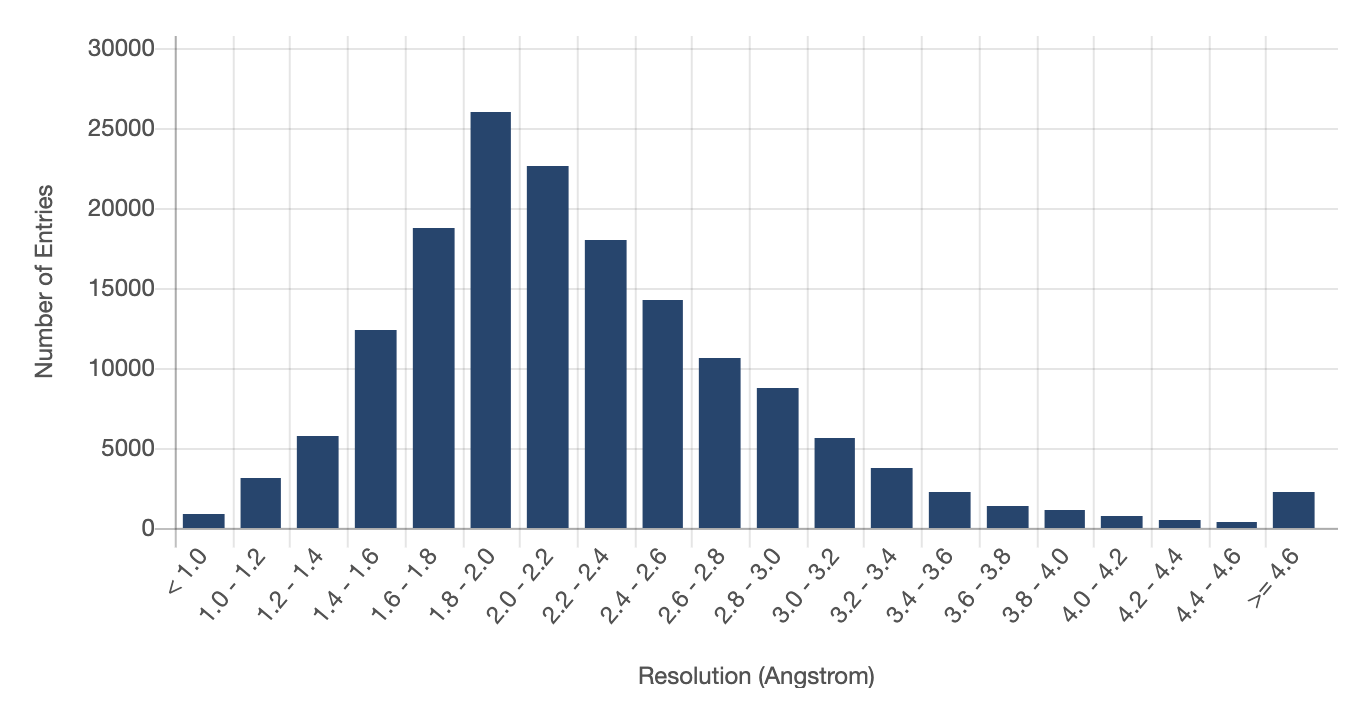
Постулируемая DeepMind оценка точности — около 1.6 Å, что, судя по графику, даже лучше, чем большинство экспериментальных структур. И здесь, наверное, можно было бы остановиться в комментировании этого пункта, но есть один нюанс. Указанная оценка точности справедлива только для \(C_{\alpha}\) атомов, тогда как на графике выше она для всех атомов структуры.
Что это значит? Честно говоря, не знаю. У человечества есть некоторые успехи в достраивании полной структуры белка на основе одной лишь структуры основного каркаса (как оптимизационные, так и на основе всё того же машинного обучения). Но никто еще не пробовал объединить AlphaFold с этими методами и оценить точность. Так что мы просто понятия не имеем какова реальная точность AlphaFold (а точнее, методов с его использованием) в получении структур, с которыми можно работать.
Что говорят оптимисты
Комментировать оптимистов гораздо менее интересно, поскольку многие из них — люди, к структурной биологии (и вообще какой-либо) отношения не имеющие, а потому высказывания там, скорее, эмоциональные, чем по делу. Я отобрал несколько тех, что мне показались значимыми.
Отрыв от второго места в 2.5 раза!
Мне кажется, мы достаточно подробно разобрали то, как устроены попугаи, в которых сравниваются алгоритмы. Лично мне из этого стало понятно только то, что я не могу никак объективно делать выводы на основе этих сравнений.
Гораздо более показательной мне кажется картинка из всё того же пресс-релиза, в которой сравниваются результаты разных лет. По ней тоже видно, что прогресс значителен. И это при том, что задачи ежегодно становятся всё сложнее и сложнее.
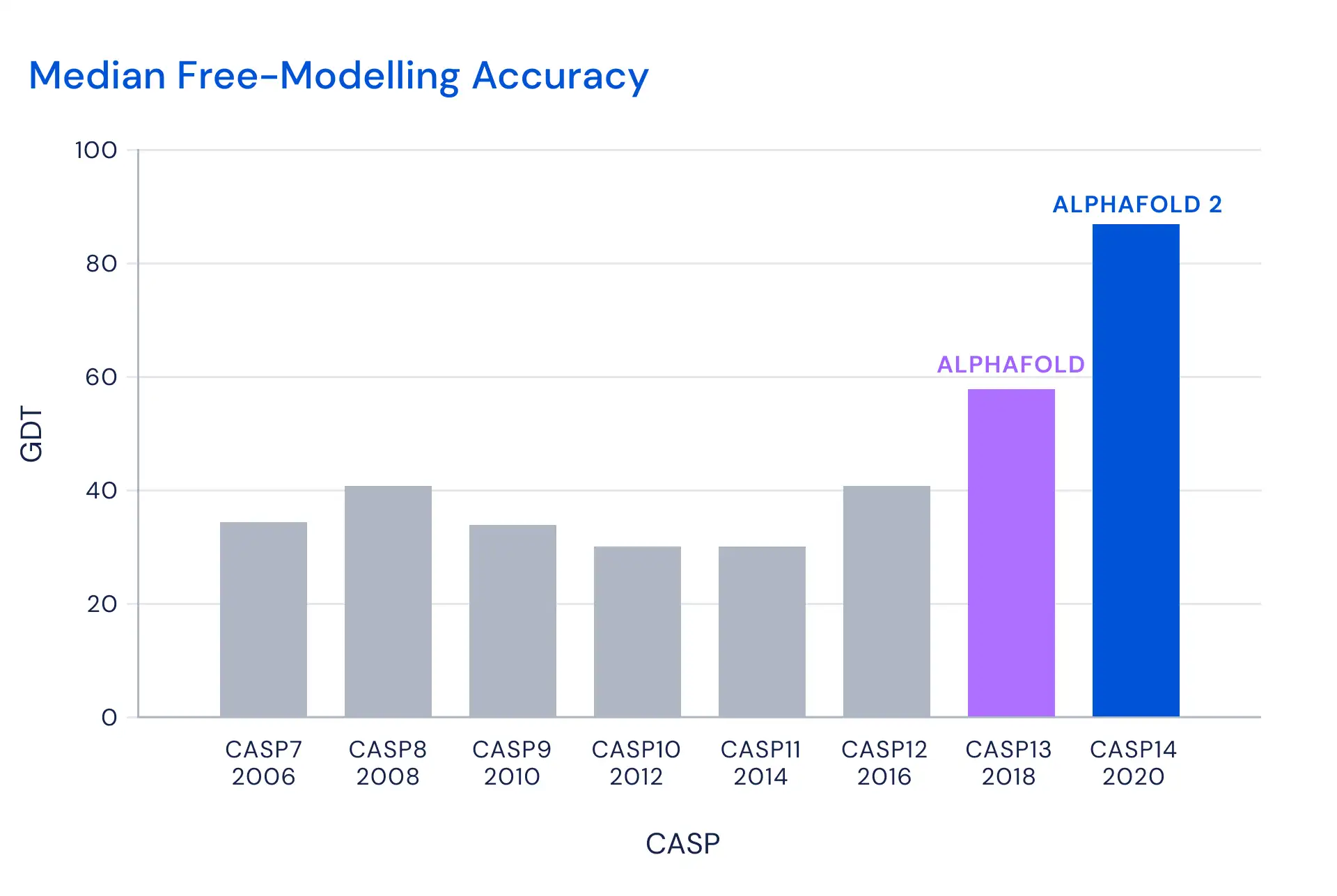
В общем, прогресс, действительно значителен. Но я бы хотел посмотреть на оценки GDT_TS для других решений, чтоб понять, может быть, они не столь отстают, как кажется, когда смотришь на результаты манипуляций с z-score.
Порой и грубой структуры достаточно
Сложно не согласиться с тем, что грубая структура лучше, чем никакая. Но на практике я не знаю ни одной важной проблемы, которую можно было бы решить с помощью одной только структуры по \(C_{\alpha}\) атомов. Возможно, моделирование поведения белков в coarse-grained виде, когда требуется просимулировать длительный отрезок времени и посмотреть на поведение белка. Но мой личный опыт с такими моделями крайне отрицательный.
Так как больше я и не знаю, что сказать по этому поводу в силу ограниченного кругозора, присовокуплю к этому пункту уже высказанное выше наблюдение, что мы умеем превращать неполные структуры в полные. Но, как высказывалось выше, насколько хорошо это всё будет работать в связке, непонятно.
Главное — метод в основе, он много где применим
Да, да и еще раз да! Можно сказать, что я сам оптимист именно по данному пункту. Отложив в сторону практическую пользу конкретно AlphaFold в том виде, что он есть сейчас, мне кажется, что сложно отрицать то, что в нашем распоряжении появился новый интересный метод, применимость которого может быть весьма высока в различных смежных задачах.
Связь коэволюции (корреляции позиций последовательностей) с расстояниями между атомами — это мощный инструмент, хорошо ложащийся на машинное обучение. В доказательство этого тезиса за прошедшие два года с обнародования результатов первого AlphaFold, появился ряд интересных решений конкретных прикладных задач.
Важный пример для меня — это лаборатория Джеффри Грея, которая сделала отличный метод для предсказания петель антител. Очень актуальная прикладная задача для нужд современной фармацевтики, которая до сих пор решается весьма и весьма плохо. Команда Грея, к слову, забила на получение матрицы коэволюции (можно понять, последовательностей антител значительно меньше, чем белков вообще, а вариабельность этих последовательностей куда выше), но внедрила пару других трюков, которые характерны именно для антител. И всё полетело.
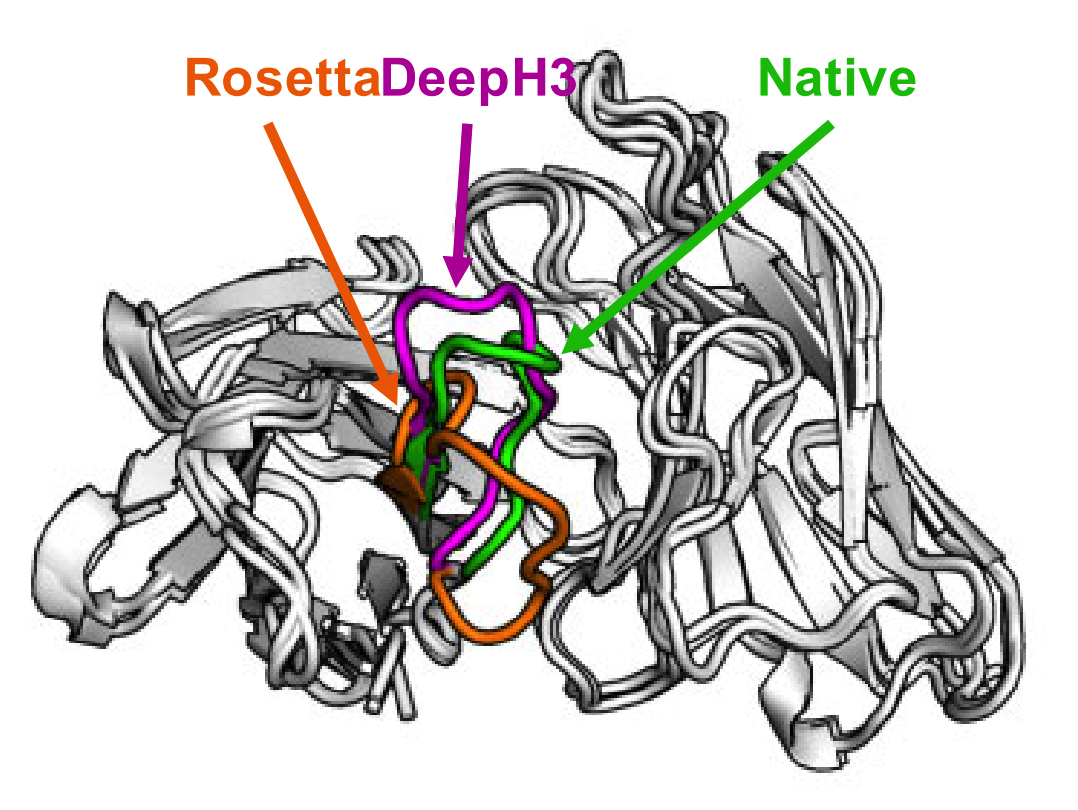
На мой взгляд, это как раз обеспечивает нам надежду на скорое появление методов для решения важных задач индустрии. Мне, как её представителю, нужно именно это, а не один алгоритм для решения фундаментальной задачи, результат работы которого я не смогу применить на практике.
Заключение, или что говорю я
Предсказание структуры белков — несомненно важный инструмент для анализа биологических систем и рационального биологического дизайна. Но нужно четко отдавать себе отчет в том, что это не серебряная пуля, а один из многих инструментов. Даже получение идеального решения этой задачи не решит все проблемы структурной биологии и кратно не удешевит/ускорит разработку новых лекарств. Но появление такого решения, несомненно, сильно продвинуло бы индустрию.
К сожалению, этого решения у нас еще нет. Даже очень качественное предсказание \(C_{\alpha}\) — это еще не предсказание структуры. Но ценность AlphaFold не в практической применимости, а в заложенной методологии, которая может быть использована при решении многих смежных задач. Так что это большой прорыв, и вся область, на мой взгляд, должна сказать DeepMind большое спасибо.
Так что впереди огромная работа по решению многих интересных задач. Специальность вычислительного биоинформатика становится все сложнее и интереснее, но и возможностей у её представителей всё больше. Пускай мы не решим все задачи одним махом, но каждую решим обязательно!